Quick Start#
Installation#
The package can be installed from PyPI using pip install pymatgen-analysis-defects.
Once installed, the different modules can be imported via the pymatgen.analysis.defects namespace.
from pymatgen.analysis.defects import core, ccd, finder
To ensure that the namespace is installed properly run
and look at the __file__ variable to make sure that it is accessing the code from pymatgen-analysis-defect and not pymatgen.
Definition of a defect#
Our code defines defects using a combination of bulk structure and sites.
Since the bulk primitive structure is, in principle, unique up to arbitrary translations, equivalence of defects can be now easily checked using StructureMatcher.
Below we show how you can create a basic substitutional defect from a pymatgen.core.Structure object by replacing one of the Ga atoms in GaN with Mg.
Note that we create a MgGa defect by creating a PeriodicSite Object mg_ga with the data of the first Ga site but
from pathlib import Path
from pymatgen.analysis.defects.core import Substitution
from pymatgen.core import PeriodicSite, Species, Structure
TEST_FILES = Path("../../../tests/test_files")
gan_struct = Structure.from_file(TEST_FILES / "GaN.vasp")
# make a substitution site
ga_site = gan_struct[0]
mg_site = PeriodicSite(
species=Species("Mg"), coords=ga_site.frac_coords, lattice=gan_struct.lattice
)
# instantiate the defect object
mg_ga = Substitution(structure=gan_struct, site=mg_site)
print(mg_ga)
Mg subsitituted on the Ga site at at site #0
mg_ga.defect_structure
Structure Summary
Lattice
abc : 3.2162901334217344 3.2162901334217344 5.239962
angles : 90.0 90.0 120.00000274450079
volume : 46.9428220153705
A : 1.608145 -2.785389 0.0
B : 1.608145 2.785389 0.0
C : 0.0 0.0 5.239962
pbc : True True True
PeriodicSite: Mg (1.608, -0.9285, 2.615) [0.6667, 0.3333, 0.4991]
PeriodicSite: Ga (Ga3+) (1.608, 0.9285, 5.235) [0.3333, 0.6667, 0.9991]
PeriodicSite: N (N3-) (1.608, -0.9285, 4.59) [0.6667, 0.3333, 0.8759]
PeriodicSite: N (N3-) (1.608, 0.9285, 1.97) [0.3333, 0.6667, 0.3759]
Instantiating FormationEnergyDiagram#
The object responsible for analyzing the formation energy is described by the following fields.
FormationEnergyDiagram(
bulk_entry: 'ComputedStructureEntry',
defect_entries: 'List[DefectEntry]',
pd_entries: 'list[ComputedEntry]',
vbm: 'float',
band_gap: 'Optional[float]' = None,
inc_inf_values: 'bool' = True,
)
Using a convenience constructor with_directories you can just feed in dictionary of {str: Path}
pointing to the directories where the supercell calculations are stored.
The keys, other than bulk will be the charge states of the calculations.
As long as the vasprun.xml and LOCPOT files are in the directories the FormationEnergyDiagram object can be constructed.
import warnings
from monty.serialization import loadfn
from pymatgen.analysis.defects.thermo import FormationEnergyDiagram
warnings.filterwarnings("ignore")
sc_dir = TEST_FILES / "Mg_Ga"
# ents = MPRester().get_entries_in_chemsys("Mg-Ga-N") # Query from MPRester
ents = loadfn(TEST_FILES / "Ga_Mg_N.json") # Load from local
fed = FormationEnergyDiagram.with_directories(
directory_map={
"bulk": sc_dir / "bulk_sc",
0: sc_dir / "q=0",
-1: sc_dir / "q=-1",
1: sc_dir / "q=1",
},
defect=mg_ga,
pd_entries=ents,
dielectric=10,
)
---------------------------------------------------------------------------
ImportError Traceback (most recent call last)
Cell In[4], line 11
7
8 sc_dir = TEST_FILES / "Mg_Ga"
9 # ents = MPRester().get_entries_in_chemsys("Mg-Ga-N") # Query from MPRester
10 ents = loadfn(TEST_FILES / "Ga_Mg_N.json") # Load from local
---> 11 fed = FormationEnergyDiagram.with_directories(
12 directory_map={
13 "bulk": sc_dir / "bulk_sc",
14 0: sc_dir / "q=0",
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/thermo.py:450, in FormationEnergyDiagram.with_directories(cls, directory_map, defect, pd_entries, dielectric, vbm, **kwargs)
443 q_entry, q_locpot = _read_dir(q_dir)
444 q_d_entry = DefectEntry(
445 defect=defect,
446 charge_state=int(qq),
447 sc_entry=q_entry,
448 )
--> 450 q_d_entry.get_freysoldt_correction(
451 defect_locpot=q_locpot,
452 bulk_locpot=bulk_locpot,
453 dielectric=dielectric,
454 )
455 def_entries.append(q_d_entry)
456 if vbm is None:
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/thermo.py:148, in DefectEntry.get_freysoldt_correction(self, defect_locpot, bulk_locpot, dielectric, defect_struct, bulk_struct, **kwargs)
143 raise ValueError(
144 msg,
145 )
147 if self.sc_defect_frac_coords is None:
--> 148 finder = DefectSiteFinder()
149 defect_fpos = finder.get_defect_fpos(
150 defect_structure=defect_struct,
151 base_structure=bulk_struct,
152 )
153 self.sc_defect_frac_coords = defect_fpos
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/finder.py:61, in DefectSiteFinder.__init__(self, symprec, angle_tolerance)
59 if SOAP is None:
60 msg = "dscribe is required to use DefectSiteFinder. Install with ``pip install dscribe``."
---> 61 raise ImportError(
62 msg,
63 )
64 self.symprec = symprec
65 self.angle_tolerance = angle_tolerance
ImportError: dscribe is required to use DefectSiteFinder. Install with ``pip install dscribe``.
Getting the formation energy diagram#
The chemical potential limits can be access using:
fed.chempot_limits
If you have crystal_toolkit and the its jupyter lab extensions installed you can view the chemical potential diagram by importing crystal_toolkit and running a cell with the the chempot_diagram on the last line.
import crystal_toolkit
fed.chempot_diagram
Note that it is different from chemical potential plot hosted on the material project because the elemental phases are set to zero energy.
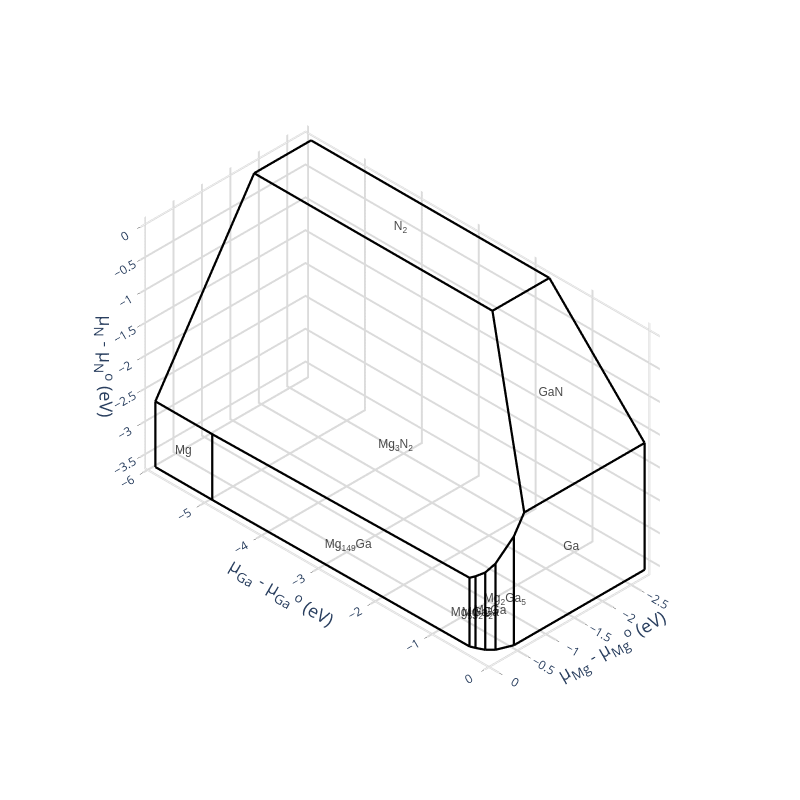
Now we can plot the formation energy diagram by calculating the Fermi level and formation energy at the different charge state transitions.
import numpy as np
from matplotlib import pyplot as plt
ts = np.array(fed.get_transitions(fed.chempot_limits[1], x_max=5))
plt.plot(ts[:, 0], ts[:, 1], "-o")
plt.xlabel("Fermi Level (eV)")
plt.ylabel("Formation Energy(eV)")