Formation Energy Diagrams#
The formation energy diagram is perhaps the most important tool in analyzing the defect properties. It informs both the likelihood of a defect forming and the transition level where the charge state of the defect changes.
The formation energy of a defect is determined by the chemical potential of everything that goes into forming the defect. While all of the different chemical potentials are interconnected, it is conceptually easier to separate them into two groups: the chemical potential of the elements and the chemical potential of the electron. The chemical potential of the electron often called the Fermi level, accounts for how all of the external conditions, including the presence of other defects, affect the thermodynamics of adding or removing an electron from the defect. Since the electrons in the system can be manipulated after the defect is formed, the Fermi level is often considered a free variable and is shown as the x-axis in the formation energy diagram. The chemical potentials of the different atoms added or removed to form the defect are less dynamic and assumed to be fixed after the defect is formed.
The expression for the formation energy of a defect is given by:
where \(E^f[X^q]\) is the formation energy of the defect, \(E_{\rm tot}[X^q]\) is the total energy of the defect, \(E_{\rm tot}[{\rm bulk}]\) is the total energy of the bulk, \(n_i = N_i^{\rm defect} - N_i^{\rm bulk}\) is the change in the number of atoms of type \(i\) between the defect and bulk supercells (positive for added atoms, negative for removed atoms), \(\mu_i\) is the chemical potential of atom type \(i\), \(q\) is the charge of the defect, \(E_{\rm F}\) is the Fermi level, and \(\Delta^q\) is finite-size correction which we have to add to account for image effects of simulating the defect in a periodic simulation cell.
A schematic of the formation energy diagram for the \(q=0\) and \(q=-1\) charge states of a hypothetical defect is shown below.
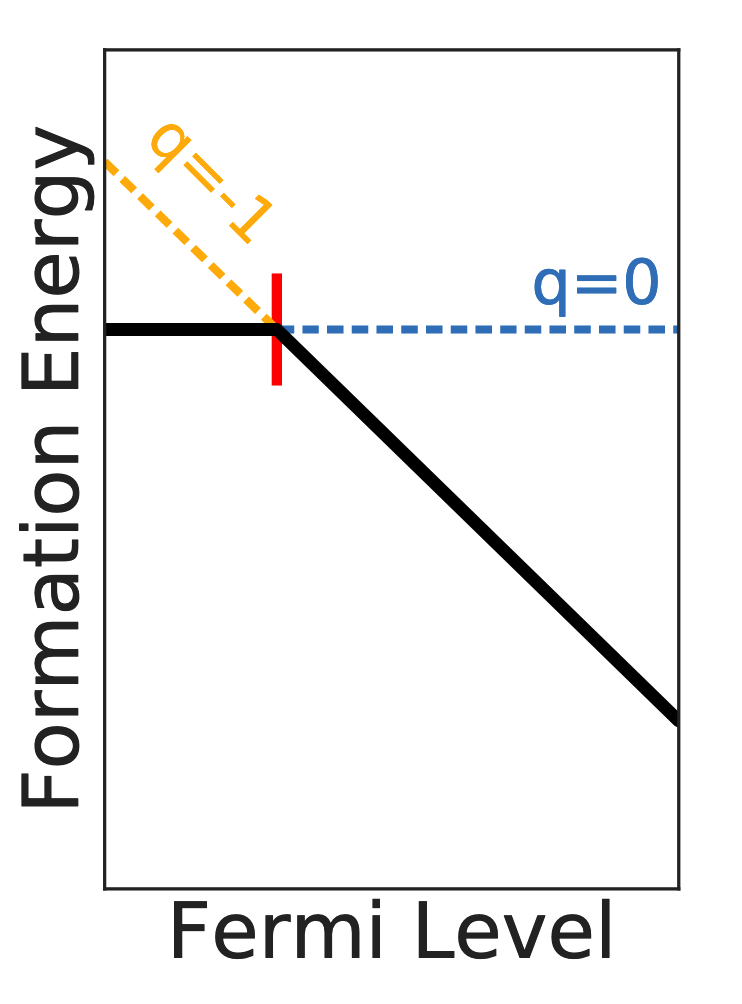
Defining a Formation Energy Diagram#
The class responsible for analyzing the formation energy is described by the following fields.
FormationEnergyDiagram(
bulk_entry: 'ComputedStructureEntry',
defect_entries: 'List[DefectEntry]',
pd_entries: 'list[ComputedEntry]',
vbm: 'float',
band_gap: 'Optional[float]' = None,
inc_inf_values: 'bool' = False,
)
Building pd_entries from your own DFT runs#
pd_entries is a list of ComputedEntry (or ComputedStructureEntry) objects describing the bulk crystal and any competing phases that bound the chemical-potential limits. Two common ways to populate it are:
From the Materials Project —
MPRester().get_entries_in_chemsys([...])returns a ready-to-use list.From local VASP runs — build a
ComputedStructureEntrydirectly from eachvasprun.xml:
from pymatgen.io.vasp import Vasprun
pd_entries = [
Vasprun(d / "vasprun.xml").get_computed_entry()
for d in [
"path/to/Mg_Ga/vasp_run",
"path/to/Mg5Ga/vasp_run",
"path/to/MgGa/vasp_run",
"path/to/Mg3N2/vasp_run",
"path/to/GaN/vasp_run",
"path/to/Mg_metal/vasp_run",
"path/to/Ga_metal/vasp_run",
"path/to/N2_molecule/vasp_run",
]
]
You can mix both sources — e.g. take pd_entries from MP and overwrite individual phases with locally computed values. The list does not need to include every entry along the convex hull; only the bulk plus the phases that bound the bulk’s stability region matter for the chemical-potential limits, and FormationEnergyDiagram will rebuild the phase diagram around the bulk entry on initialization.
import warnings
from monty.serialization import loadfn
from pymatgen.analysis.defects.thermo import FormationEnergyDiagram
warnings.filterwarnings("ignore")
from pathlib import Path
from pymatgen.analysis.defects.core import Substitution
from pymatgen.core import PeriodicSite, Species, Structure
TEST_FILES = Path("../../../tests/test_files")
gan_struct = Structure.from_file(TEST_FILES / "GaN.vasp")
# make a substitution site
ga_site = gan_struct[0]
mg_site = PeriodicSite(
species=Species("Mg"), coords=ga_site.frac_coords, lattice=gan_struct.lattice
)
# instantiate the defect object
mg_ga = Substitution(structure=gan_struct, site=mg_site)
sc_dir = TEST_FILES / "Mg_Ga"
# ents = MPRester().get_entries_in_chemsys("Mg-Ga-N") # Query from MPRester
ents = loadfn(TEST_FILES / "Ga_Mg_N.json") # Load from local
fed = FormationEnergyDiagram.with_directories(
directory_map={
"bulk": sc_dir / "bulk_sc",
0: sc_dir / "q=0",
-1: sc_dir / "q=-1",
1: sc_dir / "q=1",
},
defect=mg_ga,
pd_entries=ents,
dielectric=10,
)
---------------------------------------------------------------------------
ImportError Traceback (most recent call last)
Cell In[1], line 27
23
24 sc_dir = TEST_FILES / "Mg_Ga"
25 # ents = MPRester().get_entries_in_chemsys("Mg-Ga-N") # Query from MPRester
26 ents = loadfn(TEST_FILES / "Ga_Mg_N.json") # Load from local
---> 27 fed = FormationEnergyDiagram.with_directories(
28 directory_map={
29 "bulk": sc_dir / "bulk_sc",
30 0: sc_dir / "q=0",
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/thermo.py:450, in FormationEnergyDiagram.with_directories(cls, directory_map, defect, pd_entries, dielectric, vbm, **kwargs)
443 q_entry, q_locpot = _read_dir(q_dir)
444 q_d_entry = DefectEntry(
445 defect=defect,
446 charge_state=int(qq),
447 sc_entry=q_entry,
448 )
--> 450 q_d_entry.get_freysoldt_correction(
451 defect_locpot=q_locpot,
452 bulk_locpot=bulk_locpot,
453 dielectric=dielectric,
454 )
455 def_entries.append(q_d_entry)
456 if vbm is None:
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/thermo.py:148, in DefectEntry.get_freysoldt_correction(self, defect_locpot, bulk_locpot, dielectric, defect_struct, bulk_struct, **kwargs)
143 raise ValueError(
144 msg,
145 )
147 if self.sc_defect_frac_coords is None:
--> 148 finder = DefectSiteFinder()
149 defect_fpos = finder.get_defect_fpos(
150 defect_structure=defect_struct,
151 base_structure=bulk_struct,
152 )
153 self.sc_defect_frac_coords = defect_fpos
File ~/work/pymatgen-analysis-defects/pymatgen-analysis-defects/pymatgen/analysis/defects/finder.py:61, in DefectSiteFinder.__init__(self, symprec, angle_tolerance)
59 if SOAP is None:
60 msg = "dscribe is required to use DefectSiteFinder. Install with ``pip install dscribe``."
---> 61 raise ImportError(
62 msg,
63 )
64 self.symprec = symprec
65 self.angle_tolerance = angle_tolerance
ImportError: dscribe is required to use DefectSiteFinder. Install with ``pip install dscribe``.
Getting the Elemental Chemical Potentials#
All of the chemical potentials are, in principle, free variables that indicate the conditions of the surrounding environment while the defect is formed. These free variables are bound by the enthalpies of formation of the various compounds that share the same elements as the defect and bulk material. These competing compounds essentially establish the boundaries of the allowed chemical potentials, while the VBM and CBM of the bulk material determine the allowed range of the Fermi level. As such, first-principles calculations can be used to determine the limits of the various chemical potentials. As an example, the elemental chemical potentials for the Mg\(_{\rm Ga}\) defect in GaN are shown below:
Here since the defect must form in GaN the chemical potentials are pinned by the plane defined by the enthalpy of formation of GaN.
The limits imposed by the competing compounds are shown by the additional inequalities.
The points of interest are usually vertex points in the constrained chemical potential space so we report the full set of vertex points in FormationEnergyDiagram.chempot_limits.
for i, p in enumerate(fed.chempot_limits):
print(f"Limits for the chemical potential changes for point {i}")
for k, v in p.items():
print(f"Δμ_{k} = {v:.2f} eV")
Finite-size corrections#
Finite-size corrections are necessary to account for the fact that we are simulating a charged defect in a periodic simulation cell. The standard approach was developed by Freysoldt and co-workers and is described in the following paper:
Freysoldt, C., Neugebauer, J., & Van de Walle, C. G. (2009). Fully Ab Initio Finite-Size Corrections for Charged-Defect Supercell Calculations. Phys Rev Lett, 102(1), 016402. doi: 10.1103/PhysRevLett.102.016402
This method requires calculating the the long range term from the Coulomb interaction and a short range term from the electrostatic potential in the LOCPOT file.
While the final result of the finite-size correction is just a single number \(\Delta^q\) for each charge state, the intermediate results can still be analyzed.
This is still useful, since the defect position is automatically determined from the LOCPOT files alone, to check the planar-averaged potential plots to make sure that the defect is indeed in the correct position.
This will be evident if the planar-averaged potential difference is peaked at the two ends of the plot since the automatically determined defect position is chosen as the origin of the plot.
The short range contribution is set to zero far away from the defect, which is accomplished by average the planar electrostatic potential far away from the defect in the sampling region shown below.
The intermediate analysis (planar-averaged potentials) for calculating the Freysoldt correction for each of the the three lattice directions is shown below.
from pymatgen.analysis.defects.corrections.freysoldt import plot_plnr_avg
plot_data = fed.defect_entries[1].corrections_metadata["freysoldt"]["plot_data"]
plot_plnr_avg(plot_data[0], title="Lattice Direction 1")
plot_plnr_avg(plot_data[1], title="Lattice Direction 2")
plot_plnr_avg(plot_data[2], title="Lattice Direction 3")